“Atualmente, os pacientes contam com apenas dois tipos de medicamentos – os inibidores de acetilcolinerastase e a memantina – para tratamento. Mas nenhum dos dois realmente funciona. Com a nossa pesquisa, abre-se uma grande porta para o desenvolvimento de novos medicamentos, com possibilidade de alterar o curso da doença. As perspectivas são bastante promissoras.” diz o neurocientista William Klein, da Universidade Northwestern (Estados Unidos), que desenvolve estudos que apontam o surgimento de resistência à insulina nos neurônios de pacientes com mal de Alzheimer, o que tem levado muitos pesquisadores a associarem a doença a uma nova forma de diabetes, que afetaria especificamente o cérebro.
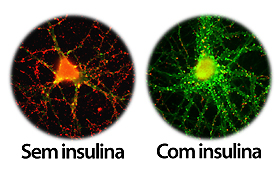 À esquerda, o neurônio doente, com sinapses reduzidas, depois de exposto a oligômeros tóxicos (pontos vermelhos). À direita, o neurônio tratado com insulina, mesmo exposto aos oligômeros, permanece saudável, com sinapses preservadas.
À esquerda, o neurônio doente, com sinapses reduzidas, depois de exposto a oligômeros tóxicos (pontos vermelhos). À direita, o neurônio tratado com insulina, mesmo exposto aos oligômeros, permanece saudável, com sinapses preservadas.
A doença de Alzheimer pode ser chamado de "diabetes" cérebro? Dois artigos recentes lançam luz sobre a relação entre estas duas doenças. Por um lado, o risco de desenvolver doença de Alzheimer é aumentada pelo aparecimento de diabetes, especialmente se o último apareceu antes da idade de 65 anos. Por outro lado, a insulina, um hormônio chave que reduz a taxa de açúcar no sangue, teria um papel protetor sobre as conexões entre as células nervosas envolvidas na memória.
Xu Wieili (Instituto Karolinska, em Estocolmo) e uma equipe sueca-americana relataram, na edição de janeiro da revista Diabetes, os resultados de um estudo realizado em mais de 13 mil gêmeos - a importância deste tipo de pesquisa que está sendo eliminar os fatores genéticos. Destes gêmeos, 467 tinham uma demência, incluindo 292 casos da doença de Alzheimer, e quase 1.400 eram diabéticos.
Os resultados sobre os pares discordantes de gêmeos mostram que o início precoce - antes dos 65 anos - da diabetes tipo 2, onde há produção de insulina, mas é ineficaz, multiplica o risco de demência comparado ao diabetes apresentado após os 65 anos. "Os fatores genéticos e ambientais podem contribuir para a associação entre diabetes e demência de início tardio, mas o ambiente (alimentação e estilo de vida) pode ser responsável na associação entre diabetes em uma idade média e demência ", concluíram os autores do artigo.
Já nos laboratórios de Doenças Neurodegenerativas e de Neurobiologia da Doenca de Alzheimer do Instituto de Bioquímica Médica da Universidade Federal do Rio de Janeiro (UFRJ), os pesquisadores descobriram que a administração da insulina em neurônios, associada à rosiglitazona. Ambos medicamentos já são empregados no tratamento do diabetes tipo 2 e podem ser poderosas armas no combate à DA, que ainda não tem cura.
Testes conduzidos pela bióloga e neurocientista Fernanda De Felice, sob o comando do professor Sérgio Teixeira Ferreira, também bioquímico e neurocientista, revelaram que as substâncias têm a propriedade de evitar a degeneração dos neurônios danificados e de restaurar sua capacidade de realizar as sinapses.
Nos últimos cinco anos, os pesquisadores começaram a relacionar a doença de Alzheimar ao diabetes tipo 2. Várias evidências clínicas mostravam que os pacientes com Alzheimer apresentavam grandes tendência a desenvolver o diabetes tipo 2 e vice-versa. Mas os fatores que relacionavam as duas doenças permaneciam obscuros. As primeiras pistas surgiram em 2008, com uma pesquisa desenvolvida por Fernanda de Felice durante um estágio na Universidade Northwestern, em Illinois, Estados Unidos. Fernanda descobriu que os neurônios dos pacientes de Alzheimer perdiam receptores de insulina. A toxina escondida no interior da célula bloqueava os neuroreceptores, impedindo que a insulina chegasse à parede do neurônio.
Até agora, não havia certeza de que o cérebro necessitasse de insulina para seu funcionamento. Na sequência, cientistas brasileiros e americanos começaram a tratar os neurônios afetados com uma combinação de insulina e roziglitazona. Hoje, graças a essas descobertas, já existe um consenso na comunidade científica de que a doença de Alzheimer pode ser, na verdade, uma espécie de diabetes do cérebro. Ferreira explica que a insulina é importantíssima para seu bom funcionamento, ajudando na obtenção de energia e na formação da memória.
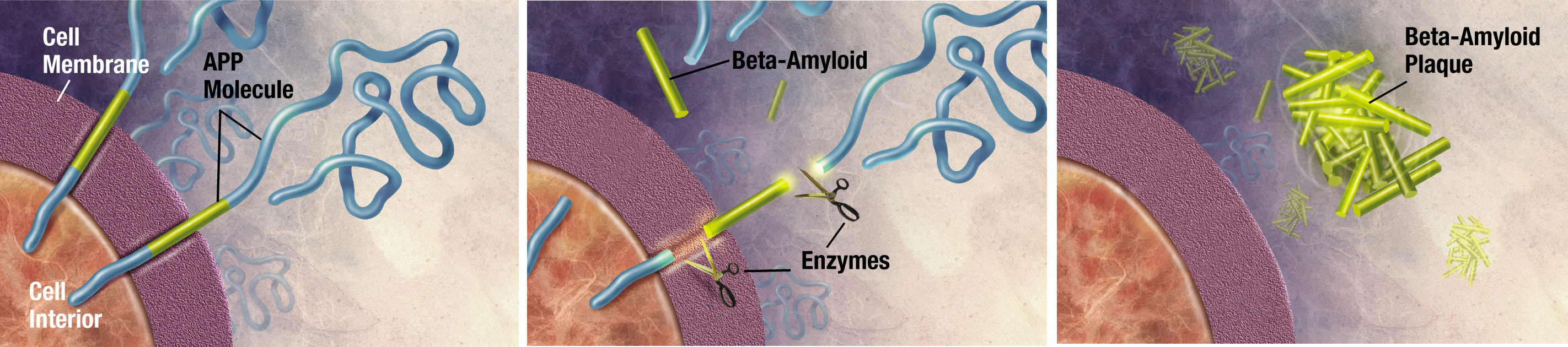
Nos portadores da doença, os neurônios se tornam resistentes à ação benéfica da insulina, daí a constatação que o mal de Alzheimer seria o diabetes tipo 3, que, ao contrário dos outros, não seria uma doença sistêmica, mas uma moléstia que ataca exclusivamente o cérebro. No processo, toxinas derivadas da proteína beta-amiloide produzidas pelo sistema nervoso central, os oligômeros, atacam os neurônios comprometendo suas funções e sua sobrevivência. Os oligômeros já são figuras conhecidas no mundo da neurociência. A novidade, segundo a pesquisa brasileira, é que não importa a composição química deles, mas, sim, sua forma. Os neuroreceptores interpretam a forma esférica da toxina como substância “autorizada”, permitindo a invasão.
Para melhor entendimento, vale lembrar que o cérebro possui um sistema de defesa próprio, que isola o sangue que irriga sua rede capilar do resto do corpo, a barreira hematoencefálica. Dessa maneira, as proteínas beta-amiloides, por serem reconhecidas como substâncias “autorizadas”, conseguem burlar a barreira e se alojar no interior dos neurônios sob a forma de oligômeros ou em emaranhados proteicos no espaço entre eles.
A experiência
Utilizando uma proteína (a lisozima) retirada da clara do ovo de galinha, a equipe sintetizou no laboratório uma estrutura semelhante aos oligômeros que atacam o cérebro. Incubada em alta temperatura e em ambiente com Ph ácido, a lisozima assumiu o formato do oligômero em menos de 24 horas. Células sadias mantidas em cultura foram expostas à ação dos oligômeros com formato de minúsculas bolinhas. Os pesquisadores observaram que, apesar de inofensiva ao organismo humano, a lisozima em forma de oligômero provocou a morte das células quando adicionada às culturas de neurônios.
A descoberta confirmou a tese de que o neurônio reconhece a forma e não a composição química do oligômero. Uma vez ligado à célula, o oligômero danifica a proteína tau, o que provoca a formação de emaranhados no seu interior e causa a deformação e a morte da célula.
O dano induzido pelos pesquisadores em células sadias ocorre poucas horas após a exposição aos oligômeros. Mas, ao aplicar a combinação da insulina e da rosiglitazona, a sensibilidade das células à insulina aumenta e a ação das duas substâncias impede que os oligômeros se liguem aos neurônios, evitando a perda de suas funções. Os neurônios submetidos à terapia tiveram as sinapses preservadas e permaneceram ativos.

A grande questão seria encontrar uma maneira de administrar o medicamento de forma que ele seja absorvido apenas pelo cérebro. Por se tratarem de substâncias que agem no sistema endócrino, seriam absorvidas por ele e apenas uma pequena quantidade chegaria ao cérebro. Por isso, a aplicação por via usual seria problemática. A administração de doses elevadas sobrecarregaria o sistema endócrino e poderia levar a um desequilíbrio na glicemia. Por outro lado, em pacientes com diabetes tipo 2, o uso continuado da insulina acaba tornando resistente a barreira hematoencefálica, em condições normais bastante permeável ao medicamento. Tal resistência agravaria a situação dos neurônios atingidos pela ação dos oligômeros.
Uma das soluções imaginadas seria produzir uma forma inalante da substância. Absorvido pela mucosa nasal, o medicamento venceria com maior facilidade a barreira hematoencefálica e chegaria mais rapidamente ao cérebro.
Dito e feito! Os resultados preliminares (bastante otimistas) dos testes podem ser conferidos nessa reportagem do Estadão.
Bibliografia:















{kind=link}
{kind=link}